

Regulation of New Medical Devices 'Fatally Flawed': Report
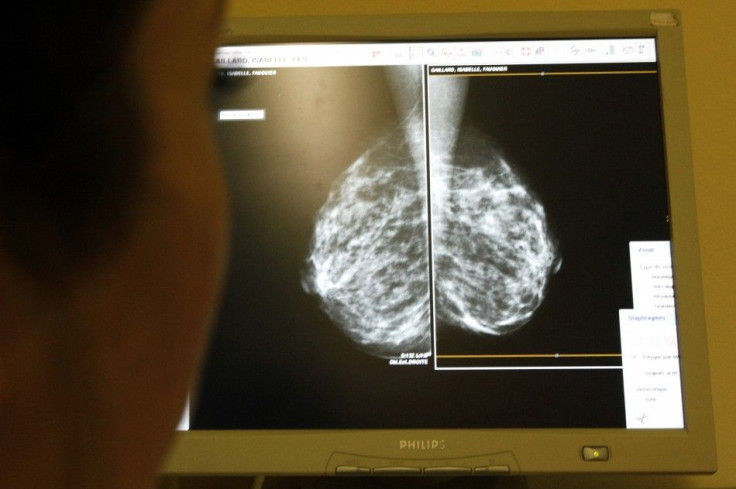
New medical devices that are hazardous or ineffective have made it onto the market because of a regulatory loophole, a report by the U.S. Institute of Medicine charged.
The Food and Drug Administration is responsible for vetting a range of products, from bandages to pacemakers, to ensure their safety and effectiveness before they are available to consumers. But a process known as 510(k) exempts devices that are "substantially equivalent" to previous products from a rigorous review, weakening the FDA's ability to properly screen the products, the report said.
"The 510(k) process generally is not intended to evaluate the safety and effectiveness of medical devices and, furthermore, cannot be transformed into a premarket evaluation of safety and effectiveness," the report's authors wrote in a letter to the FDA.
The authors of the report, which the FDA commissioned in 2009, recommended that the 501(k) framework be altered given what Dr. David Challoner called "fatal flaws" in the process. The report pointed out that some new devices are approved for being "substantially equivalent" to prior devices that had never been examined for safety, and maintained that it was unclear whether 501(k) had inhibited or encouraged medical innovation. In 2009 vast majority of devices under FDA's regulatory jurisdiction, more than 90%, were cleared under 510(k), according to the report.
A massive recall of more than 90,000 artificial hips last summer underscored concerns raised in the report, with about 1 in 8 recipients needing replacements for hips that could release small metallic particles into patients' bloodstreams over time.
Consumer rights groups have criticized the 501(k) process, saying that it favors the medical industry by allowing them to rush new products to market.
"We believe that the process for approving devices under the 510(k) program frequently is deficient because of the application and the loose interpretation of what it means to be "substantially equivalent to a predicate device," said Michael Carome, deputy director of Public Citizen's health research group.
Representatives of the medical industry, by contrast, warned against cumbersome rules that would prevent them from delivering new forms of treatment.
"We remain concerned about efforts to overhaul a regulatory pathway that would create additional uncertainties and slow patient access to medical therapies," Mark Leahey, president of the Medical Device Manufacturers Association, said. "Moving forward, we will continue to work with FDA and all stakeholders to ensure a more predictable and reasonable process is in place."
© Copyright IBTimes 2024. All rights reserved.
-

Senate Prevents Potential Air Travel Chaos, Reauthorizes FAA For Five-Years
-

Panama Papers Law Firm Boss Ramon Fonseca Dead
-

Pandemic Agreement Talks Go To The Wire
-
Low-cost MRI Paired With AI Produces High-quality Results
-

School's Out: How Climate Change Threatens Education
-

Hong Kong Demands Online Platforms Remove Banned Protest Song
-

2023 'Year Of Record Climatic Hazards' In Latin America: UN
-

Crypto Community Buzzes As Grayscale Withdraws Ethereum Futures ETF Application
-

Mongolia's Wildlife At Risk From Overgrazing
-

Milking Venom From Australia's Deadly Marine Animals









